Two-State Model
The two-state multimodal model is a quantum mechanical model for absorption spectra. It expects two states and any number of modes.
This model represents a system with two states, a ground state and an excited state. The absorption spectrum is generated by first expanding a vibronic hamiltonian. Then, the hamiltonian is diagonalized, and the eigenvalues and eigenvectors are used to calculate the spectrum peaks. Finally, the peaks are broadened with a linear combination of gaussian distributions, returning the absorption spectrum.
More details on the vibronic hamiltonian portion of this process can be seen in the Two-State Computation Docs.
More details on the peak points and peak broadening portion of this process can be seen in the Hamiltonian Computation Docs.
Model Name
In config files, this Model is named two-state. This can be specified in your-config.toml with:
model.name = "two-state"
In the package, this Model is named TwoStateModel. This can be imported with:
from quantumspectra_2024.absorption import TwoStateModel
Parameters
All Models have a mixture of required and optional parameters. For further explanation and expected values, see the Two-State Computation Docs.
Model Parameters
Note
All Two-State Models require these parameters on initialization.
These scalar parameters are used in the broadening process:
These scalar parameters are used in the vibronic hamiltonian expansion:
These vectory parameters are used in the vibronic hamiltonian expansion. They must be inputted as arrays, with one value for each mode:
General Parameters
All Models have optional parameters to specify the range of their generated spectrum. By default, all Models generate 2,001 points between 0 and 20,000 wavenumbers.
Examples
These examples display both types of usage for this class. Both use the same “default” parameters that appear in the given sample config.
Both of these methods will produce the following absorption spectrum:
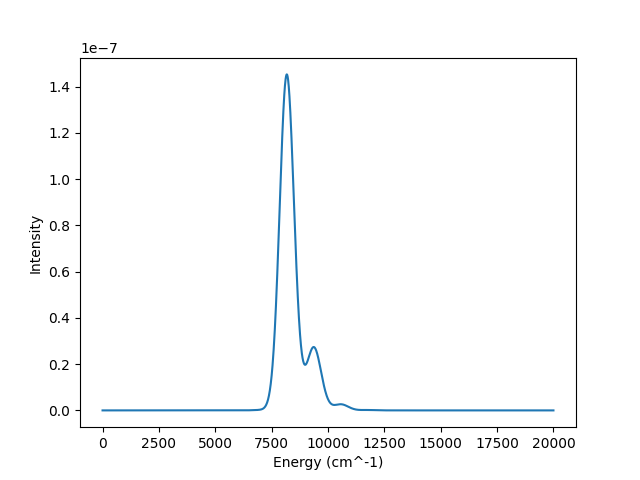
CLI Usage
First, create a config file
your-config.tomlthat includes the required specifications, and these contents:model.name = "two-state" model.temperature_kelvin = 300 model.broadening = 200 model.transfer_integral = 100 model.energy_gap = 8000 model.mode_basis_sets = [20, 200] model.mode_frequencies = [1200, 100] model.mode_couplings = [0.7, 2.0]
More information about config files can be found on the Config Files page. Sample config files can be found in the Sample Configs section of that page.
Then, run the absorption spectrum command with the path to your config file.
qs_2024 path/to/your-config.tomlThis will save the generated absorption spectrum to the specified output file.
Package Usage
First, import the Model.
from quantumspectra_2024.models import TwoStateModel
Create an instance of the model with the desired parameters.
model = TwoStateModel( temperature_kelvin=300, broadening=200, transfer_integral=100, energy_gap=8000, mode_basis_sets=[20, 200], mode_frequencies=[1200, 100], mode_couplings=[0.7, 2.0] )
Run the
get_absorptionmethod to generate the absorption spectrum.Warning
This is the cpu (or gpu)-intensive part of the process. Only run this method when you are ready to generate the absorption spectrum.
spectrum = model.get_absorption()
This will return an
AbsorptionSpectruminstance to thespectrumvariable. Details on theAbsorptionSpectrumclass can be found in the Absorption Spectrum Docs.Accessing spectrum data:
x, y = spectrum.energies, spectrum.intensities print(x) print(y)
Saving spectrum data:
spectrum.save_data("path/to/output/file.csv") spectrum.save_plot("path/to/output/plot.png")
Full Class
- class quantumspectra_2024.models.TwoStateModel(*, start_energy: float = 0.0, end_energy: float = 20000.0, num_points: int = 2001, broadening: float = 200.0, temperature_kelvin: float, transfer_integral: float, energy_gap: float, mode_basis_sets: Int[Array, 'num_modes'], mode_frequencies: Float[Array, 'num_modes'], mode_couplings: Float[Array, 'num_modes'])[source]
A two-state quantum mechanical model for absorption spectra.
- Parameters:
- start_energyfloat
absorption spectrum’s starting energy (wavenumbers).
- end_energyfloat
absorption spectrum’s ending energy (wavenumbers).
- num_pointsint
absorption spectrum’s number of points (unitless).
- broadening: float
absorption spectrum broadening factor (wavenumbers).
- temperature_kelvinfloat
system’s temperature (Kelvin).
- transfer_integralfloat
transfer integral between the two states.
- energy_gapfloat
energy gap between the two states (wavenumbers).
- mode_basis_setsFloat[Array, “num_modes”]
basis set size per mode (unitless).
- mode_frequenciesFloat[Array, “num_modes”]
frequency per mode (wavenumbers).
- mode_couplingsFloat[Array, “num_modes”]
excited state coupling per mode.
Methods
apply_electric_field(field_strength, ...)Applies an electric field to the model.
Compute the absorption spectrum for the model.
Returns the model's associated HamiltonianModel.
- get_absorption() AbsorptionSpectrum[source]
Compute the absorption spectrum for the model.
First computes the Hamiltonian, then diagonalizes it to get eigenvalues and eigenvectors. Then computes the absorption spectrum peaks and broadens them into a spectrum.
See docs in
quantumspectra_2024.modules.hamiltonian.HamiltonianComputationandTwoStateComputationto see how this is done.- Returns:
- AbsorptionSpectrum
the model’s parameterized absorption spectrum.
- get_hamiltonian() HamiltonianModel[source]
Returns the model’s associated HamiltonianModel.
- Returns:
- HamiltonianModel
the model’s Hamiltonian.
- apply_electric_field(field_strength: float, field_delta_dipole: float, field_delta_polarizability: float) TwoStateModel[source]
Applies an electric field to the model. Returns a new instance of the model.
- Parameters:
- field_strengthfloat
the strength of the electric field.
- field_delta_dipolefloat
the change in dipole moment due to the electric field.
- field_delta_polarizabilityfloat
the change in polarizability due to the electric field.
- Returns:
- TwoStateModel
the model with the electric field applied.